Outras Doenças de Plasmócitos
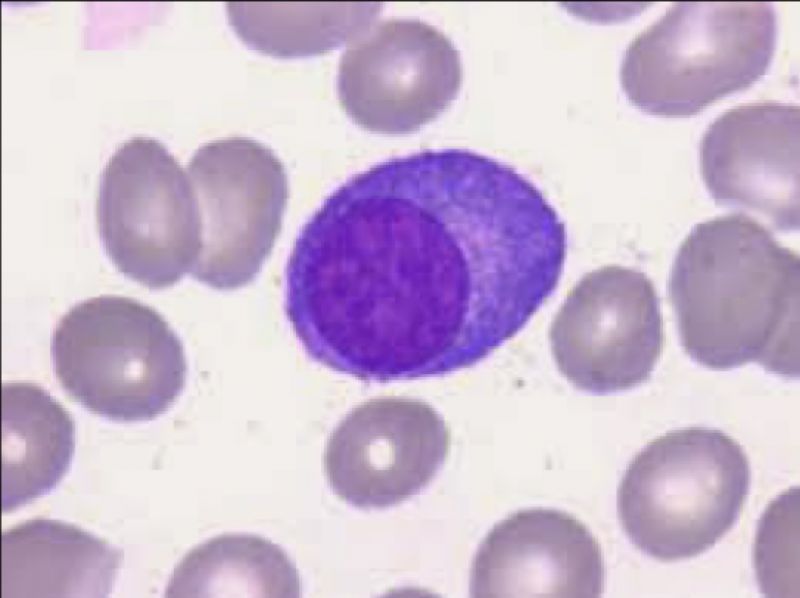
O quadro laboratorial do Mieloma Múltiplo é clássico, e cursa com Plasmócitos e Rouleaux eritrocitário principalmente. Entretanto a presença de plasmócitos não é exclusividade do MM. Vejamos outras situações que acometem estas células:
Plasmocitoma:
Tumor maligno, originado da proliferação irreversível e autônoma dos plasmócitos, podendo se apresentar como massa circunscrita ou infiltração difusa. Incomum antes dos 30 anos. Tem predomínio no sexo masculino e sua principal localização é a coluna vertebral. No plasmocitoma solitário, a eletroforese de proteínas séricas, o mielograma e as análises laboratoriais e radiológicas não apresentam evidências de doença sistêmica.
A leucemia de células plasmáticas:
A LCP é uma desordem linfoproliferativa muito rara. Seu diagnóstico é realizado com a presença de > 20% de plasmócitos no sangue periférico ou > de 2000 plasmócitos/mm³ com imunofenotipagem confirmando clonalidade. A leucemia de células plasmáticas pode ser primária, quando ocorre sem o antecedente de mieloma múltiplo (MM) ou secundária quando é uma evolução rara e agressiva do MM. A maior incidência ocorre na sexta década de vida em homens e afrodescendentes.
Macroglobulinemia de Waldenström:
A MW é uma doença linfoproliferativa dos linfócitos B, caracterizada por um linfoma linfoplasmocítico na medula óssea e por hipergamaglobulinemia monoclonal de tipo IgM. Tem um percurso clínico normalmente indolente, atingindo principalmente os indivíduos com idades entre 63 e 68 anos. A maioria dos doentes apresenta sintomas e manifestações clínicas relacionadas com a hiperviscosidade, resultante da gamopatia monoclonal IgM e/ou com as citopenias, resultantes da infiltração medular pelo linfoma.
Doença de Cadeia Pesada:
São distúrbios tipicamente malignos dos plasmócitos. Na maioria dos distúrbios dos plasmócitos, as M-proteínas são estruturalmente similares às moléculas dos anticorpos normais. Por outro lado, nas doenças de cadeia pesada, as imunoglobulinas monoclonais incompletas são produzidas. Elas consistem apenas em componentes das cadeias pesadas (alfa [α], gama [γ], mu [μ] ou delta [δ]) sem cadeias leves). A maior parte das proteínas de cadeias pesadas é de fragmentos dos seus correspondentes normais com deleções internas de extensão variável; essas deleções parecem resultar de mutações estruturais. O quadro clínico se parece mais com linfoma do que com mieloma múltiplo. As doenças de cadeia pesada são consideradas em pacientes com manifestações clínicas que sugiram distúrbios linfoproliferativos.
Amiloidose:
Consiste em grupo de doenças que possuem em comum o depósito extracelular de substância constituída principalmente por proteínas do tipo amiloide. A proteína amiloide possui propriedades ultraestruturais e bioquímicas específicas de modo que, sob métodos específicos de coloração, é possível demonstrar seu caráter fibrilar e caracterizar sua estrutura e modo de disposição nos tecidos nos quais ela se deposita. A presença física dos depósitos de substância amiloide pode ocasionar disfunção dos tecidos e órgãos onde esteja, por interação das fibrilas com receptores locais e por citotoxicidade dos depósitos. A apresentação clínica varia conforme o órgão e tecido acometido; pode ser sutil e inespecífica, como fadiga, perda de peso, até mais grave, com insuficiência renal, cardíaca, gastrintestinal e neuropatia periférica autonômica e/ou sensitivo-motora. Em geral, embora vários órgãos possam estar acometidos, uma das formas pode predominar. A amiloidose pode ser primária, hereditária ou estar associada a várias situações clínicas; e se não estiver associada a qualquer outra enfermidade é primária. A forma secundária da doença ocorre principalmente associada às neoplasias, doenças inflamatórias ou infecciosas crônicas.
Plasmocitoma:
Tumor maligno, originado da proliferação irreversível e autônoma dos plasmócitos, podendo se apresentar como massa circunscrita ou infiltração difusa. Incomum antes dos 30 anos. Tem predomínio no sexo masculino e sua principal localização é a coluna vertebral. No plasmocitoma solitário, a eletroforese de proteínas séricas, o mielograma e as análises laboratoriais e radiológicas não apresentam evidências de doença sistêmica.
A leucemia de células plasmáticas:
A LCP é uma desordem linfoproliferativa muito rara. Seu diagnóstico é realizado com a presença de > 20% de plasmócitos no sangue periférico ou > de 2000 plasmócitos/mm³ com imunofenotipagem confirmando clonalidade. A leucemia de células plasmáticas pode ser primária, quando ocorre sem o antecedente de mieloma múltiplo (MM) ou secundária quando é uma evolução rara e agressiva do MM. A maior incidência ocorre na sexta década de vida em homens e afrodescendentes.
Macroglobulinemia de Waldenström:
A MW é uma doença linfoproliferativa dos linfócitos B, caracterizada por um linfoma linfoplasmocítico na medula óssea e por hipergamaglobulinemia monoclonal de tipo IgM. Tem um percurso clínico normalmente indolente, atingindo principalmente os indivíduos com idades entre 63 e 68 anos. A maioria dos doentes apresenta sintomas e manifestações clínicas relacionadas com a hiperviscosidade, resultante da gamopatia monoclonal IgM e/ou com as citopenias, resultantes da infiltração medular pelo linfoma.
Doença de Cadeia Pesada:
São distúrbios tipicamente malignos dos plasmócitos. Na maioria dos distúrbios dos plasmócitos, as M-proteínas são estruturalmente similares às moléculas dos anticorpos normais. Por outro lado, nas doenças de cadeia pesada, as imunoglobulinas monoclonais incompletas são produzidas. Elas consistem apenas em componentes das cadeias pesadas (alfa [α], gama [γ], mu [μ] ou delta [δ]) sem cadeias leves). A maior parte das proteínas de cadeias pesadas é de fragmentos dos seus correspondentes normais com deleções internas de extensão variável; essas deleções parecem resultar de mutações estruturais. O quadro clínico se parece mais com linfoma do que com mieloma múltiplo. As doenças de cadeia pesada são consideradas em pacientes com manifestações clínicas que sugiram distúrbios linfoproliferativos.
Amiloidose:
Consiste em grupo de doenças que possuem em comum o depósito extracelular de substância constituída principalmente por proteínas do tipo amiloide. A proteína amiloide possui propriedades ultraestruturais e bioquímicas específicas de modo que, sob métodos específicos de coloração, é possível demonstrar seu caráter fibrilar e caracterizar sua estrutura e modo de disposição nos tecidos nos quais ela se deposita. A presença física dos depósitos de substância amiloide pode ocasionar disfunção dos tecidos e órgãos onde esteja, por interação das fibrilas com receptores locais e por citotoxicidade dos depósitos. A apresentação clínica varia conforme o órgão e tecido acometido; pode ser sutil e inespecífica, como fadiga, perda de peso, até mais grave, com insuficiência renal, cardíaca, gastrintestinal e neuropatia periférica autonômica e/ou sensitivo-motora. Em geral, embora vários órgãos possam estar acometidos, uma das formas pode predominar. A amiloidose pode ser primária, hereditária ou estar associada a várias situações clínicas; e se não estiver associada a qualquer outra enfermidade é primária. A forma secundária da doença ocorre principalmente associada às neoplasias, doenças inflamatórias ou infecciosas crônicas.
Gostou do nosso conte�do?
Cadastre-se para receber nossos conte�dos exclusivos.


